Protein O-mannosylation in development and physiology
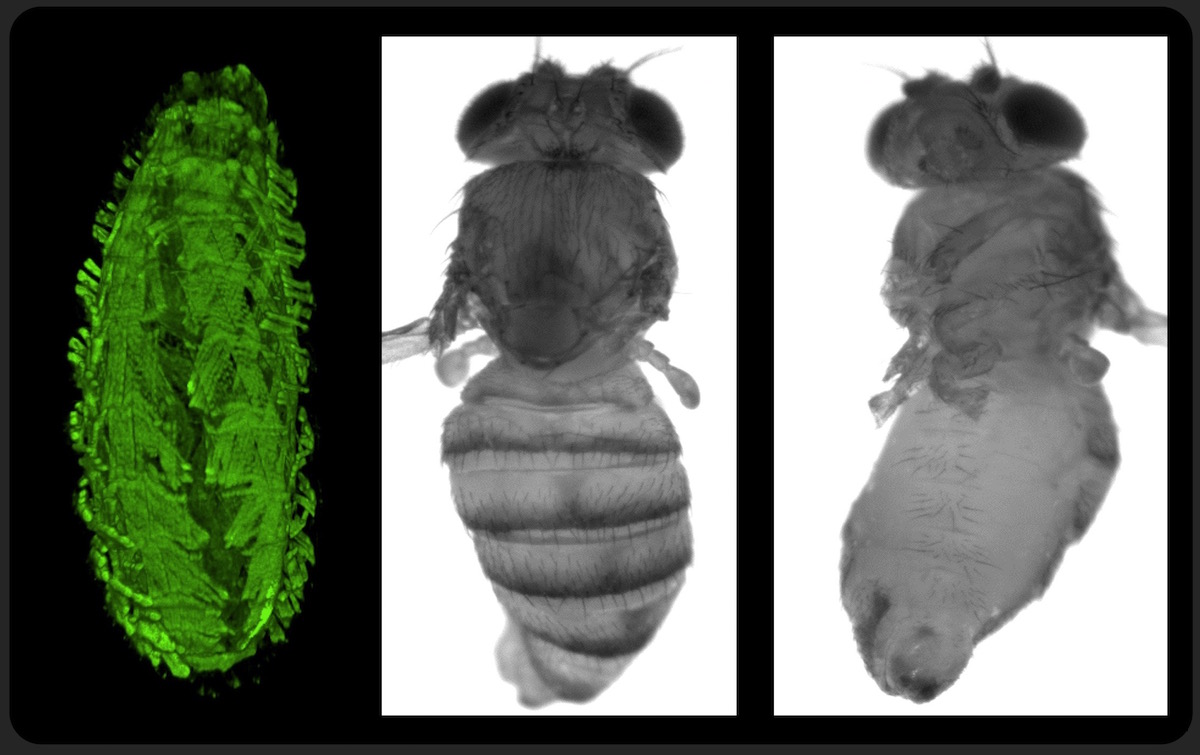
|
POMT mutants have body torsion phenotype which reveals the chirality of pathogenic mechanisms.
Left panel: embryo with fluorescently labeled muscles. Right panels: adult fly (wings and legs removed) |
Compromised protein O-mannosylation is associated with severe muscular dystrophies and defects of the nervous system. The O-mannosylation pathway is evolutionarily conserved in all animals, however its molecular and genetic mechanisms are complex and not well understood. The best-characterized target of O-mannosylation is Dystroglycan (DG), a glycoprotein central to the assembly of the dystrophin-glycoprotein complex (DGC) in the sercolemma of muscle cells. DGC provides a bridge between cytoskeleton and the extracellular matrix (ECM), which is essential for muscle physiology and the integrity of muscle plasma membrane. An enzymatic complex of two protein O mannosyltransferases, POMT1 and POMT2 attaches O-linked mannose to serine and threonine residues of alpha-Dystroglycan, an extracellular part of DG. O-mannosyl glycans play an essential part in interactions between DG and its extracellular matrix ligands, such as laminin and neurexin. Compromised biosynthesis of O-mannosyl glycans on DG results in severe congenital muscular dystrophies termed dystroglycanopathies, including Walker Warburg syndrome (WWS) and Muscle-Eye-Brain disease (MEB).
In all metazoan organisms, O-mannosylation is mediated by a complex of two protein O–mannosyltransferases (POMTs), POMT1 and POMT2. In Drosophila, POMT1 and POMT2 correspond to Rotated Abdomen (RT) and Twisted (TW), respectively, the genes with a characteristic mutant phenotype of misalignment (“rotation”) of abdominal segments in adult flies. Functional properties of POMT1 and POMT2 are well conserved between Drosophila and mammals. We established that RT and TW can modify Drosophila DG in vitro and in vivo. In collaboration with mass spectrometry experts, we characterized O‑glycosylation of DG, which unveiled a surprising abundance of O-mannosyl glycans within its mucin-type domain. These experiments also identified a new O-mannosylated region of DG and indicated that the acceptor specificity of O-mannosyltransferases is complex, and their substrate recognition is determined not simply by a local consensus sequence but also depends on acceptor properties on a larger structural scale. Our results discovered an interplay between O-mannosylation and O-GalNAc glycosylation pathways at the level of acceptor site modifications. This suggests that phenotypes of POMT mutants can result from ectopic O-GalNAc modifications that take place in the absence of O-linked mannose, which potentially represents a new pathogenic mechanism associated with defects in O-mannosylation. In our current research, we continue combining genetic and molecular approaches in order to understand how O-mannosylation regulates development and physiology of muscles and the nervous system, while also focusing on characterizing new functionally relevant O–mannosylated glycoproteins.
Publications
Baker R, Nakamura N, Chandel I, Howell B, Lyalin D, and Panin VM (2018) Protein O-Mannosyltransferases Affect Sensory Axon Wiring and Dynamic Chirality of Body Posture in the Drosophila Embryo. J Neurosci. 38(7):1850-1865.
Panin VM and Wells L (2014) Protein O-mannosylation in metazoan organisms. Protocols in Protein Science 75: Unit 12.12.
Nakamura N, Lyalin D, Panin VM. (2010) Protein O-mannosylation in animal development and physiology: From human disorders to Drosophila phenotypes. Semin Cell Dev Biol. 21(6): 622-630.
Nakamura N, Stalnaker S, Lyalin D, Lavrova O, Wells L, Panin V. (2010) Drosophila Dystroglycan is a target of O‑mannosyltransferase activity of two protein O‑mannosyltransferases, Rotated Abdomen and Twisted. Glycobiology. 20:381-94.
Lyalin, D.A., Koles, K., Roosendaal, S.D., Repnikova, E. A., Van Wechel, L., and Panin, V. M. (2006) The twisted gene encodes Drosophila protein O‑mannosyltransferase 2 and genetically interacts with the rotated abdomen gene encoding Drosophila protein O-mannosyltransferase 1. Genetics. 172(1): 343-53.